Microbiome data is leading to innovative solutions in diverse industries, from human and animal health to agriculture and the built environment. Next-generation sequencing has allowed researchers new insights into the microbial world with high levels of resolution—that is, they can precisely identify many of the bacteria and other microorganisms present. Not only that, but these technologies have enabled higher throughput than ever before. Foundational technologies, such as amplification and sequencing of phylogenetic markers, including the 16S rRNA gene, have become standard tools for understanding how microbial communities are structured and how they respond to changes in their environment.
However, amplicon sequencing does have some limitations in the type and resolution of the information it provides. This is where metagenomics — the direct recovery of total genomic information from the environment — can make a difference. Amplicon sequencing readily provides information at roughly the genus level; with care, it can identify microbial species and strains only under specific circumstances. Metagenomics reliably provides up to strain-level resolution (Figure 1). It also provides information about function—what the microorganisms’ genes equip them to do.
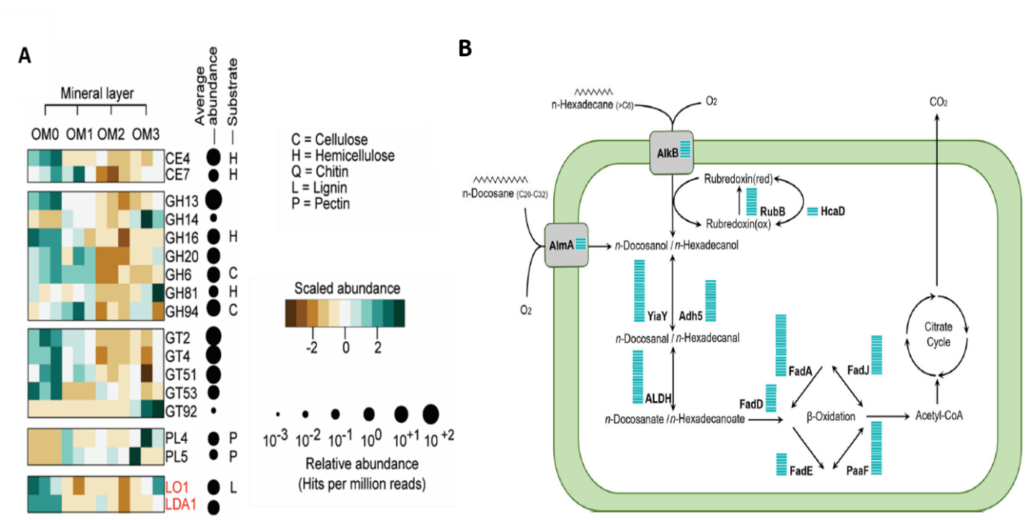
Functional information is useful to understand the mechanisms underlying the changes in the microbial community, to reconstruct the metabolism of the community as an entity, and to discover new genes and pathways (Figure 2). The addition of functional information is also helpful to understand what groups provide what functions and how much redundancy exists for that function, which can have implications for the degree of resilience of the community (how it can bounce back after perturbations).
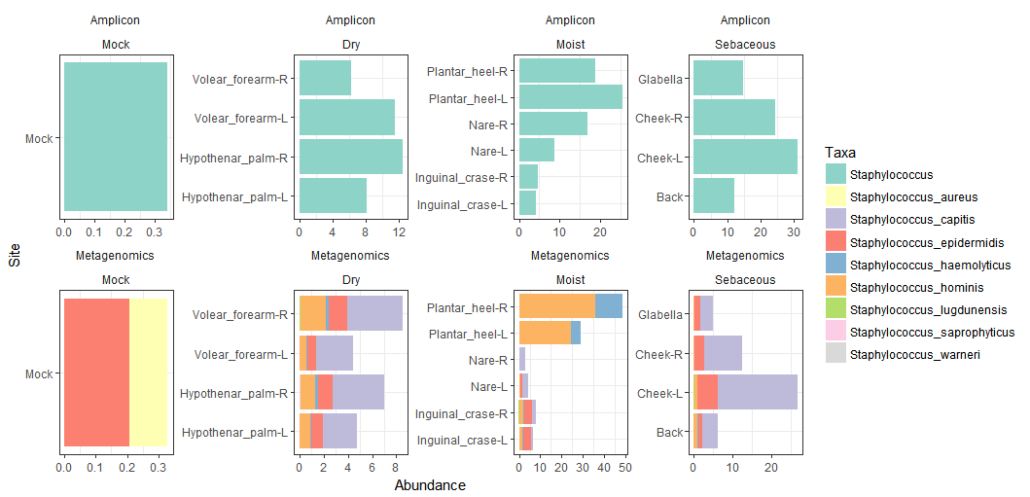
A second advantage of metagenomics is that it recovers data from all microbial community members, so the information will not be limited to bacteria (as when using a 16S rRNA) but also include data for fungi, viruses, and other groups. One example: using metagenomics, Oh et al. 2014 (Figure 3) mapped the abundance of bacterial and fungal species, and viral groups to different skin locations, identified functional gene differences across sites, and recovered 67 partial genomes (bacterial, viral, and eukaryotic). When samples have low diversity (e.g. enrichments), metagenomics can recover high quality draft genome sequences from community members. The genome of Kuenenia stuttgardiensis, one of the first characterized anaerobic ammonia oxidizers, was obtained from a metagenome of a bioreactor sample (see doi:10.1038/nature04647) without the need for cultivation.
Metagenomics also comes with its own limitations. Since sequencing is done for the whole community, analysis can be challenging if too much host DNA is present or for samples with very low biomass. In the first case, most of the data will be of little interest since the host is not the target. In the second case, only a small part of the community will be reflected in the data, leading to a biased understanding of the microbiome. Finally, the applications of metagenomics depend on the depth of sequencing (Figure 4). Having higher sequencing coverage allows for recovery of data from more community members, assembly of short reads into larger contigs, and the use of those contigs to reconstruct genes, pathways, and genomes.
In addition to these challenges, the public databases which are used for data comparison are constrained. These databases contain sequence information as well additional data such as the organism the sequences came from, the location and date of sampling, functional annotation, and links to related publications. Databases link sequence information with taxonomy and function and represent the historic efforts of researchers worldwide (and consequently their biases). These databases are limited first because most genes in any genome, even those from well-studied groups, lack biochemical characterization; and second, databases are biased towards human-related and pathogenic groups. Poorly represented groups in the databases include the archaea, fungi, viruses, and small eukaryotes; poorly represented environments include soils. Yet, this may not be a roadblock, but a challenge that will lead us to a better understanding of the microbial world.
"Both the cost and complexity barriers to metagenomic and metatranscriptomic sequencing have been greatly reduced, meaning these shotgun approaches are now practical ways to very precisely profile the human microbiome and other microbial communities," says Curtis Huttenhower, Microbiome Insights Scientific Advisory Board member and Associate Professor of computation biology and bioinformatics at the Harvard T.H. Chan School of Public Health (Boston). "Metagenomics can now easily provide strain tracking and functional information that is difficult to obtain using amplicon sequencing, and these can further be integrated with metatranscriptomics, metabolomics, or other culture-independent molecular data to understand microbial community bioactivity."
Microbiome Insights provides a full suite of services, including both amplicon sequencing and metagenomics. We can help you answer the question: where will metagenomics can take you?
Are you planning a microbiome study? We created this guide to help.